Fabry disease (also called Anderson-Fabry disease, AFD, or Fabry’s disease) is a rare genetic condition that was discovered a century ago. Approximately 1 in 40,000 people have the disease. In the past, treatment was limited to symptom management. Clinical trials of recently-developed enzyme replacement therapies have shown to be safe and effective in slowing the progression of the disease and improving the quality of life for people living with Fabry.
Fabry disease is an inherited disorder that affects the body’s ability to metabolize or break down fatty substances called lipids. One of these lipids, globotriaosylceramide (GL-3), requires the enzyme called alpha-galactosidase A (alpha-GAL A) to be broken down. In those with Fabry disease, alpha-GAL A is either missing or the enzyme is faulty. Consequently, GL-3 is not removed from the blood and accumulates in body tissues and organs. That’s why Fabry disease is usually referred to as a storage disorder.
Two chromosomes determine the sex of a person: males have one X and one Y chromosome, while females have two X chromosomes. The gene that causes the missing or faulty alpha-GAL A enzyme is located on the X chromosome. Here is a look at how the disease is passed from parent to child:
If the parent with the disease is the father, he will pass it on to all of his daughters but none of his sons, because the father passes on the affected X chromosome to the daughters and the Y chromosome to the sons.
If the parent with the disease is the mother, there is a 50-50 chance she will pass it on to each child, regardless of the child’s sex, because the mother only passes on X chromosomes to her children, one of which carries Fabry disease.
Over time, the abnormal storage of GL-3 lipids increases to harmful levels. GL-3 buildup in blood vessel walls narrows the vessels and decreases blood flow, affecting the kidneys, heart, brain, skin, and nervous system. The eyes can also be affected. Females may show fewer signs of the disorder, because they have two X chromosomes, one of which is healthy. However, males with Fabry inherit only the faulty X chromosome and can experience all aspects of the illness. Symptoms include:
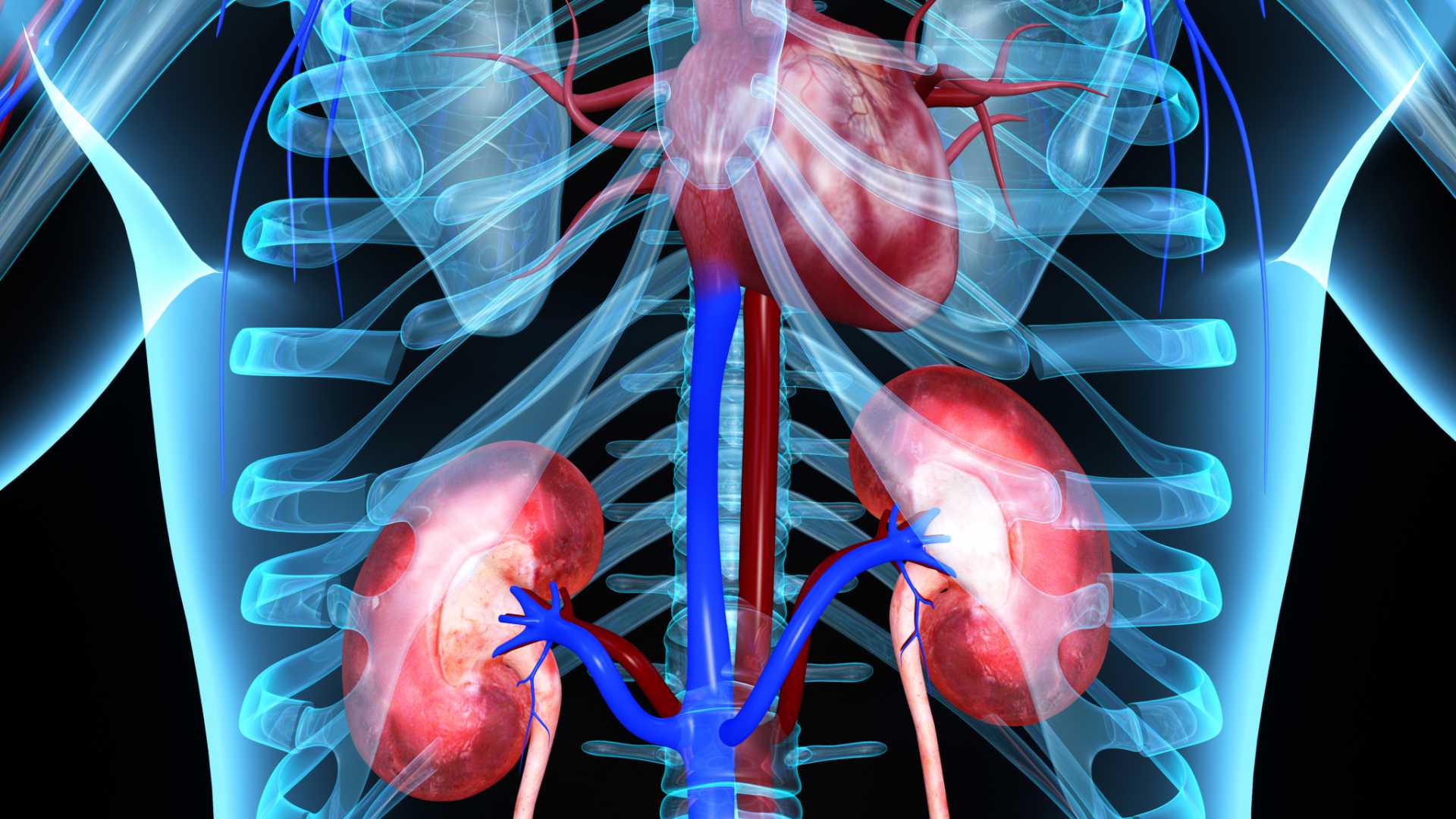
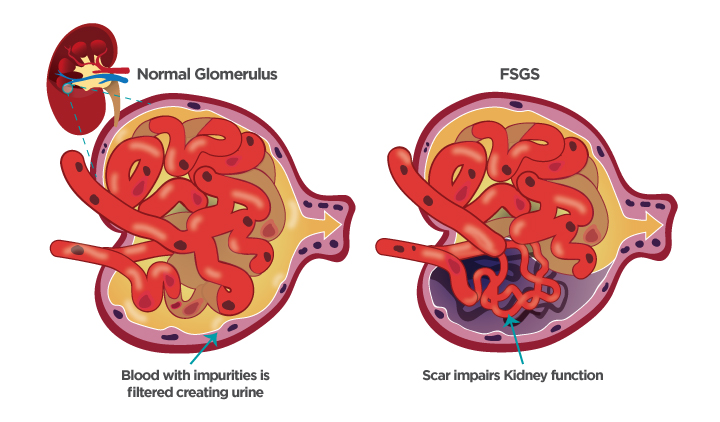
Within the kidneys, the accumulation of GL-3 causes glomerulonephritis with proteinuria and a gradual loss of kidney function, which can lead to end stage renal disease (ESRD). People with ESRD need dialysis or a kidney transplant in order to live.
Although symptoms of Fabry disease usually appear during childhood or early adolescence, diagnosing the illness may be difficult since many of the symptoms are also experienced with other disorders. In general, the more severe symptoms develop gradually over time, but heart and kidney disease can occur in childhood. Early detection, treatment, and monitoring are helpful in slowing the progression of the disease. If a family member tests positive for Fabry disease, it’s recommended that immediate relatives be tested for it as well.
A probable diagnosis is made based on the symptoms exhibited by patients. A clinical diagnosis can be confirmed through an enzyme assay. This test can confirm Fabry disease by showing very low or no alpha GAL A activity in the blood, white blood cells, or tissues collected through a biopsy. It is recommended that this test be followed by a molecular genetic analysis.
The severe pain associated with Fabry disease usually responds well to anti-seizure drugs such as Dilantin® or Neurontin®. ACE inhibitors and other high blood pressure medications may help delay loss of kidney function. Heart disease can be treated with antiarrhythmic drugs, pace makers, and bypass surgery if the patient has coronary artery disease.
Enzyme replacement therapy (ERT) to treat Fabry disease specifically became available in the early 2000s. Studies have shown a significant reduction in pain; a stabilization of kidney function; reduction of GL-3 deposits in the kidneys, heart, and skin; and improvement of the clinical symptoms of the disease. However, serious adverse reactions with ERT can occur and the medication should only be administered under close medical supervision.
Although Fabry disease is rare, support systems are available for those who have it. If you have been diagnosed with the condition, check out National Fabry Disease Foundation and Fabry Support and Information Group (FSIG) for more information.
Fabry disease can be difficult to diagnose since it shares many symptoms with other illnesses and because it is rarely encountered. Making an appointment with a doctor after experiencing any symptoms of Fabry disease is the first step to treatment if it is diagnosed. A family history of Fabry disease should be considered. The earlier the disease is diagnosed, the better chance of saving kidney function and preventing other serious complications.